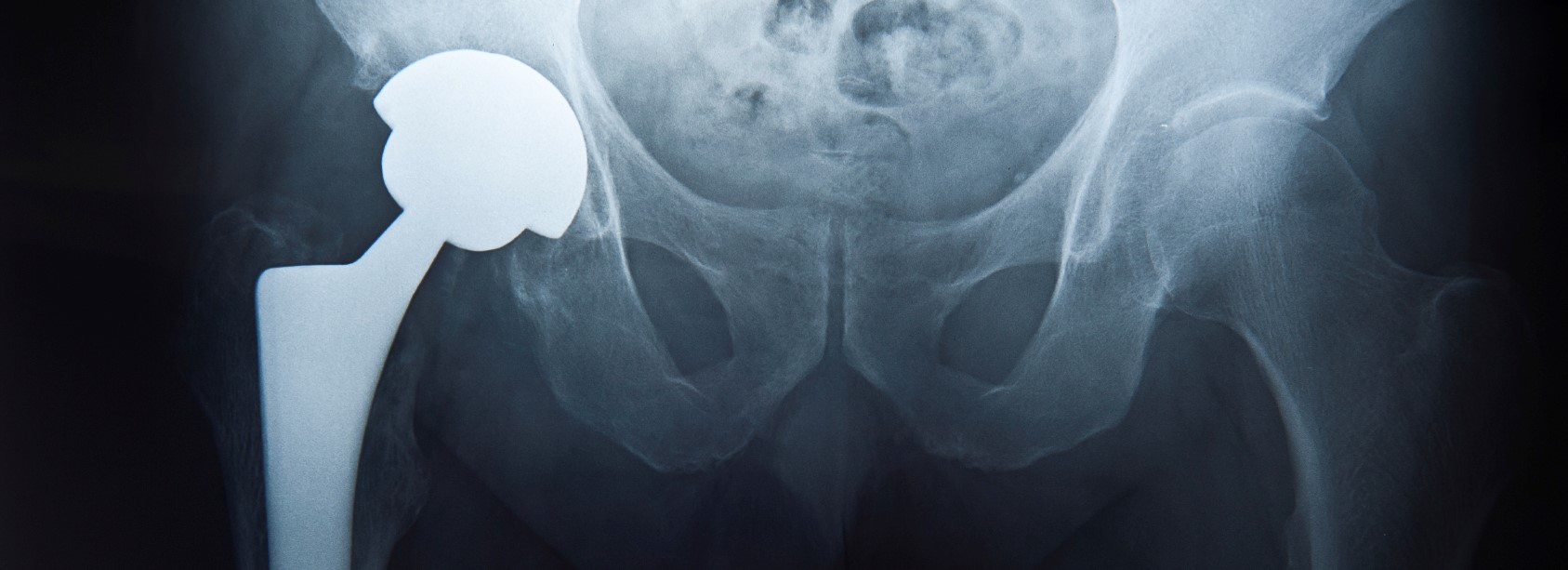
 Recently published research reveals that the Food and Drug Administration (FDA) has not done an adequate job at regulating hip replacement systems. As the first trials over Johnson & Johnson’s defective Pinnacle Ultamet hip implant equipment begin across the United States, patients and doctors are left wondering whether these manufacturers were asked to provide evidence for the safety of their products. According to a new scientific study, which looks at 50 recent implant technologies, almost 85 percent of manufacturers failed to submit evidence on the safety or effectiveness of the product. Despite this, the FDA approved the hip replacement systems for medical use in humans. Ongoing experiences with defective implant products, including
Recently published research reveals that the Food and Drug Administration (FDA) has not done an adequate job at regulating hip replacement systems. As the first trials over Johnson & Johnson’s defective Pinnacle Ultamet hip implant equipment begin across the United States, patients and doctors are left wondering whether these manufacturers were asked to provide evidence for the safety of their products. According to a new scientific study, which looks at 50 recent implant technologies, almost 85 percent of manufacturers failed to submit evidence on the safety or effectiveness of the product. Despite this, the FDA approved the hip replacement systems for medical use in humans. Ongoing experiences with defective implant products, including
Safety Testing Lacking
A common FDA approval pathway for new medical devices is the 510(k) clearance, which allows manufacturers to circumvent safety testing requirements by submitting scientific evidence that the new device is equivalent to an already-cleared product. Reports by the Institute of Medicine, however, show that out of 50 recently approved implant devices in their sample, only eight presented data to prove their similarity to existing products already on the market. Indeed, only 31 out of the 1105 technologies studied in total had complied with the standards set by the 510(k) — less than three percent. In many cases, manufacturers would present no scientific evidence at all, or would do so without summarizing the results. These products were nevertheless approved for marketing.
Neither has the FDA been very good at following up. Typically, manufacturers are supposed to submit postmarket studies that corroborate the safety of their equipment. Yet, any studies that the FDA had ordered were often too small to properly evaluate the safety of the device and companies often went unpenalized for delaying the studies. Between 2005 and 2011, a report by the PEW Charitable Trusts found that the agency ordered 223 postmarket studies for 158 high-risk medical devices; these resulted in only one device removed from the market and in 31 label changes. Experts have questioned the efficacy of label changes, since this information often doesn’t get translated to the consumer. Furthermore, some have accused the postmarket studies of suffering from possible flaws, as they are often designed in large part by the manufacturer.
Metal-on-metal hip implants, many of which were introduced to the market with a 510(k) clearance, have been under fire for defects in a growing number of brands. Stryker Corporation and DePuy, a franchise of Johnson and Johnson, are facing lawsuits for their defective Trident, Rejuvenate, ABG II, Accolade TMZF, and ASR hip replacement systems and components. Prone to undue fretting and corrosion, these products are known to cause significant pain and discomfort, as well as metallosis. Known as metal poisoning, metallosis results from the release of metal debris into the bloodstream and can lead to additional pain, as well as contribute to the heightened risk of implant failure amongst patients. DePuy and Stryker have been accused of negligence and deceptive marketing practices, since evidence shows that they were aware of the health risks associated with their products, yet failed to disclose the information when marketing the devices to doctors and patients.
Protect Your Health and Schedule Your Free Case Review
Close to 300,000 hip replacements are conducted each year in the United States. These technologies are vital to the quality of life of hundreds of thousands of patients. Consumers deserve to be well-informed about the medical devices they use and companies that withhold crucial information about their products should be penalized. If you were fitted with a metal-on-metal hip replacement system and are experiencing undue pain, contact the Georgia law offices of Pope McGlamry to schedule your complimentary case review. Our experienced legal team can help you fight for the compensation you deserve; give us a call at 404.523.7706.